



Ce sont des rapports préliminaires (preprint) qui n'ont pas fait l'objet d'un examen par les pairs. Ils ne doivent pas être considérés comme concluants, guider la pratique clinique / les comportements liés à la santé, ni être rapportés dans les médias comme des informations établies.

Mais ce rapport a néanmoins été publié par une équipe de chercheurs indiens composée de Prashant Pradhan, Ashutosh Kumar Pandey, Akhilesh Mishra, Parul Gupta, Praveen Kumar Tripathi, Manoj Balakrishnan Menon, James Gomes, Perumal Vivekanandan et Bishwajit Kundu.
Il est daté du 31 janvier 2020 et on le retrouve à l'adresse suivante : https://www.biorxiv.org/content/10.1101/2020.01.30.927871v1.full
Voici une traduction française de ce rapport intitulé « Similitude étrange d'inserts uniques dans la protéine de pointe 2019-nCoV avec le VIH-1 gp120 et Gag », qui a soulevé une vague de théories conspirationnistes.
Nous assistons actuellement à une épidémie majeure causée par le nouveau coronavirus 2019 (2019-nCoV). L'évolution de 2019-nCoV reste insaisissable. Nous avons trouvé 4 insertions dans la glycoprotéine de pointe (S) qui sont uniques au 2019-nCoV et ne sont pas présentes dans d'autres coronavirus. Il est important de noter que les résidus d'acides aminés dans les 4 inserts ont une identité ou une similitude avec ceux du VIH-1 gp120 ou du VIH-1 Gag. Fait intéressant, bien que les inserts soient discontinus sur la séquence d'acides aminés primaire, la modélisation 3D du 2019-nCoV suggère qu'ils convergent pour constituer le site de liaison du récepteur. La découverte de 4 inserts uniques dans le 2019-nCoV, qui ont tous une identité / similitude avec les résidus d'acides aminés dans les protéines structurales clés du VIH-1, est peu probable de nature fortuite. Ce travail fournit des informations encore inconnues sur le nCoV 2019 et met en lumière l'évolution et la pathogénicité de ce virus avec des implications importantes pour le diagnostic de ce virus.
Il est daté du 31 janvier 2020 et on le retrouve à l'adresse suivante : https://www.biorxiv.org/content/10.1101/2020.01.30.927871v1.full
Voici une traduction française de ce rapport intitulé « Similitude étrange d'inserts uniques dans la protéine de pointe 2019-nCoV avec le VIH-1 gp120 et Gag », qui a soulevé une vague de théories conspirationnistes.
Nous assistons actuellement à une épidémie majeure causée par le nouveau coronavirus 2019 (2019-nCoV). L'évolution de 2019-nCoV reste insaisissable. Nous avons trouvé 4 insertions dans la glycoprotéine de pointe (S) qui sont uniques au 2019-nCoV et ne sont pas présentes dans d'autres coronavirus. Il est important de noter que les résidus d'acides aminés dans les 4 inserts ont une identité ou une similitude avec ceux du VIH-1 gp120 ou du VIH-1 Gag. Fait intéressant, bien que les inserts soient discontinus sur la séquence d'acides aminés primaire, la modélisation 3D du 2019-nCoV suggère qu'ils convergent pour constituer le site de liaison du récepteur. La découverte de 4 inserts uniques dans le 2019-nCoV, qui ont tous une identité / similitude avec les résidus d'acides aminés dans les protéines structurales clés du VIH-1, est peu probable de nature fortuite. Ce travail fournit des informations encore inconnues sur le nCoV 2019 et met en lumière l'évolution et la pathogénicité de ce virus avec des implications importantes pour le diagnostic de ce virus.
Introduction
Les coronavirus (CoV) sont des virus à ARN à sens positif simple brin qui infectent les animaux et les humains. Ceux-ci sont classés en 4 genres en fonction de leur spécificité d'hôte : Alphacoronavirus, Betacoronavirus, Deltacoronavirus et Gammacoronavirus (Snijder et al., 2006). Il existe sept types de CoV connus, dont 229E et NL63 (Genus Alphacoronavirus), OC43, HKU1, MERS et SARS (Genus Betacoronavirus). Alors que 229E, NL63, OC43 et HKU1 infectent couramment les humains, les éclosions de SRAS et de MERS en 2002 et 2012 se sont produites respectivement lorsque le virus est passé des animaux aux humains, provoquant une mortalité importante (J.Chan et al., Sd; JFW Chan et al., 2015). En décembre 2019, une autre épidémie de coronavirus a été signalée à Wuhan, en Chine, qui s'est également transmise des animaux aux humains. Ce nouveau virus a été temporairement nommé nouveau coronavirus 2019 (2019-nCoV) par l'Organisation mondiale de la santé (OMS) (J.F.-W.Chan et al., 2020; Zhu et al., 2020). Bien qu'il existe plusieurs hypothèses sur l'origine du 2019-nCoV, la source de cette épidémie en cours reste difficile à cerner.
Les schémas de transmission du 2019-nCoV sont similaires aux schémas de transmission documentés lors des flambées précédentes, notamment par contact corporel ou en aérosol avec des personnes infectées par le virus. Des cas de maladie légère à sévère et des décès dus à l'infection ont été signalés à Wuhan. Cette épidémie s'est propagée rapidement dans des pays éloignés, notamment la France, l'Australie et les États-Unis. Le nombre de cas à l'intérieur et à l'extérieur de la Chine augmente fortement. Notre compréhension actuelle se limite aux séquences du génome du virus et à de modestes données épidémiologiques et cliniques. Une analyse complète des séquences 2019-nCoV disponibles peut fournir des indices importants qui peuvent aider à faire progresser notre compréhension actuelle pour gérer l'épidémie en cours.
La glycoprotéine de pointe (S) du cornonavirus est clivée en deux sous-unités (S1 et S2). La sous-unité S1 aide à la liaison aux récepteurs et la sous-unité S2 facilite la fusion membranaire (Bosch et al., 2003; Li, 2016). Les glycoprotéines de pointe des coronovirus sont des déterminants importants du tropisme tissulaire et de la gamme d'hôtes. De plus, les glycoprotéines de pointe sont des cibles critiques pour le développement de vaccins (Du et al., 2013). Pour cette raison, les protéines de pointe représentent les plus étudiées parmi les coronavirus. Nous avons donc cherché à étudier la glycoprotéine de pointe du 2019-nCoV pour comprendre son évolution, sa séquence de caractéristiques nouvelles et ses caractéristiques structurelles à l'aide d'outils de calcul.
Les schémas de transmission du 2019-nCoV sont similaires aux schémas de transmission documentés lors des flambées précédentes, notamment par contact corporel ou en aérosol avec des personnes infectées par le virus. Des cas de maladie légère à sévère et des décès dus à l'infection ont été signalés à Wuhan. Cette épidémie s'est propagée rapidement dans des pays éloignés, notamment la France, l'Australie et les États-Unis. Le nombre de cas à l'intérieur et à l'extérieur de la Chine augmente fortement. Notre compréhension actuelle se limite aux séquences du génome du virus et à de modestes données épidémiologiques et cliniques. Une analyse complète des séquences 2019-nCoV disponibles peut fournir des indices importants qui peuvent aider à faire progresser notre compréhension actuelle pour gérer l'épidémie en cours.
La glycoprotéine de pointe (S) du cornonavirus est clivée en deux sous-unités (S1 et S2). La sous-unité S1 aide à la liaison aux récepteurs et la sous-unité S2 facilite la fusion membranaire (Bosch et al., 2003; Li, 2016). Les glycoprotéines de pointe des coronovirus sont des déterminants importants du tropisme tissulaire et de la gamme d'hôtes. De plus, les glycoprotéines de pointe sont des cibles critiques pour le développement de vaccins (Du et al., 2013). Pour cette raison, les protéines de pointe représentent les plus étudiées parmi les coronavirus. Nous avons donc cherché à étudier la glycoprotéine de pointe du 2019-nCoV pour comprendre son évolution, sa séquence de caractéristiques nouvelles et ses caractéristiques structurelles à l'aide d'outils de calcul.
Méthodologie
Récupération et alignement des séquences d'acide nucléique et de protéine
Nous avons récupéré toutes les séquences de coronavirus disponibles (n = 55) dans la base de données du génome viral NCBI (https://www.ncbi.nlm.nih.gov) et nous avons utilisé le GISAID (Elbe & Buckland-Merrett, 2017) [https://www.gisaid.org] pour récupérer toutes les séquences disponibles (n = 28) de 2019-nCoV au 27 janvier 2020. L'alignement de séquences multiples de tous les génomes de coronavirus a été effectué à l'aide du logiciel MUSCLE (Edgar, 2004) basé sur la méthode de jonction des voisins. Sur 55 génomes de coronavirus, 32 génomes représentatifs de toutes les catégories ont été utilisés pour le développement d'arbres phylogénétiques à l'aide du logiciel MEGAX (Kumar et al., 2018).Le parent le plus proche s'est révélé être le CoV du SRAS. La région glycoprotéique du SARS CoV et 2019-nCoV a été alignée et visualisée à l'aide du logiciel Multalin (Corpet, 1988). La séquence d'acides aminés et de nucléotides identifiée était alignée avec la base de données du génome viral entier en utilisant BLASTp et BLASTn. La conservation des motifs de nucléotides et d'acides aminés dans 28 variantes cliniques du génome 2019-nCoV a été présentée en effectuant un alignement de séquences multiples à l'aide du logiciel MEGAX. La structure tridimensionnelle de la glycoprotéine 2019-nCoV a été générée en utilisant le serveur en ligne SWISS-MODEL (Biasini et al., 2014) et la structure a été marquée et visualisée en utilisant PyMol (DeLano, 2002).
Résultats
Similitude étrange de nouveaux inserts dans la protéine de pointe 2019-nCoV avec le VIH-1 gp120 et Gag
Notre arbre phylogentique de coronavirus pleine longueur suggère que le 2019-nCoV est étroitement lié au CoV du SRAS [Fig1]. De plus, d'autres études récentes ont établi un lien entre le nCoV 2019 et le CoV du SRAS. Nous avons donc comparé les séquences de glycoprotéines de pointe du 2019-nCoV à celle du SARS CoV (numéro d'accès NCBI : AY390556.1). Après un examen attentif de l'alignement de séquence, nous avons constaté que la glycoprotéine de pointe 2019-nCoV contient 4 insertions [Fig.2]. Pour étudier plus avant si ces inserts sont présents dans un autre virus corona, nous avons effectué un alignement de séquences multiples des séquences d'acides aminés de glycoprotéines de pointe de tous les coronavirus disponibles (n = 55) [voir le tableau S.File1] dans NCBI refseq (ncbi.nlm .nih.gov) cela inclut une séquence de 2019-nCoV [Fig.S1]. Nous avons constaté que ces 4 insertions [inserts 1, 2, 3 et 4] sont uniques au 2019-nCoV et ne sont pas présentes dans d'autres coronavirus analysés. Un autre groupe de Chine avait documenté trois insertions comparant moins de séquences de glycoprotéines à pointes de coronavirus (Zhou et al., 2020).
Alignement de séquences multiples de glycoprotéines de la famille des coronaviridae, représentant les quatre inserts.
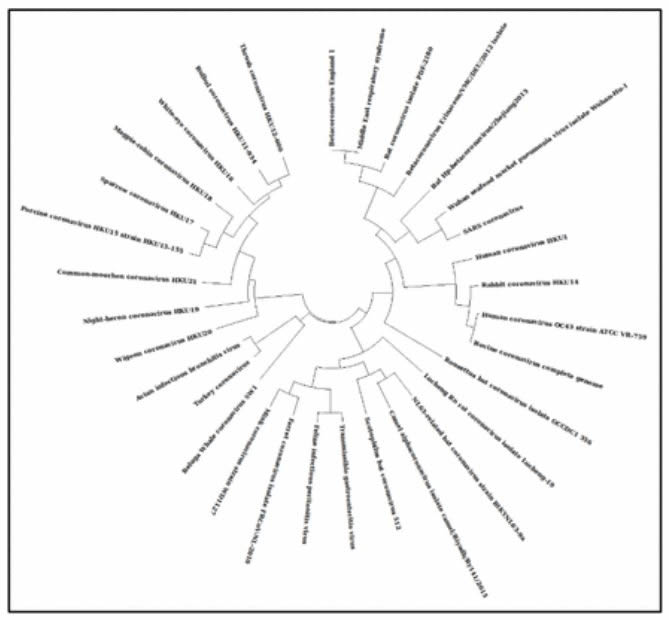
L'histoire évolutive a été déduite en utilisant la méthode du maximum de vraisemblance et le modèle basé sur la matrice JTT. L'arbre avec la plus forte probabilité logarithmique (12458.88) est montré. Les arborescences initiales pour la recherche heuristique ont été obtenues automatiquement en appliquant les algorithmes de voisinage et de BioNJ à une matrice de distances par paires estimées à l'aide d'un modèle JTT, puis en sélectionnant la topologie avec une valeur de vraisemblance logarithmique supérieure. Cette analyse a impliqué 5 séquences d'acides aminés. Il y avait un total de 1387 positions dans l'ensemble de données final. Des analyses évolutives ont été réalisées dans MEGA X.

Les séquences de protéines de pointe de 2019-nCoV (Wuhan-HU-1, Accession NC_045512) et de SARS CoV (GZ02, Accession AY390556) ont été alignées à l'aide du logiciel MultiAlin. Les sites de différence sont mis en évidence dans des encadrés.
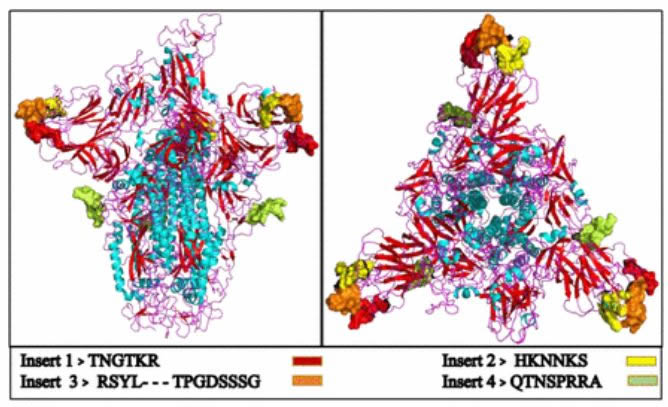
Les inserts de la protéine d'enveloppe du VIH sont représentés par des billes colorées, présentes au site de liaison de la protéine.
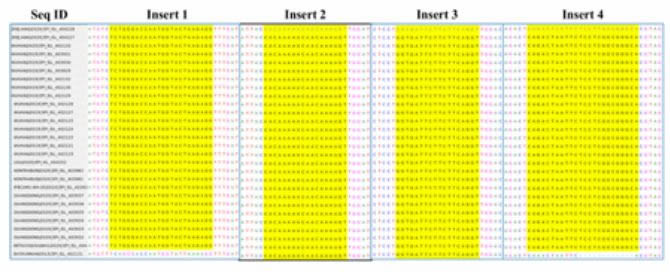
Nous avons ensuite analysé toutes les séquences disponibles (n = 28) de nCoV 2019 dans GISAID (Elbe & Buckland-Merrett, 2017) au 27 janvier 2020 pour la présence de ces inserts. Comme la plupart de ces séquences ne sont pas annotées, nous avons comparé les séquences nucléotidiques de la glycoprotéine de pointe de toutes les séquences 2019-nCoV disponibles en utilisant BLASTp. Fait intéressant, les 4 insertions ont été absolument (100%) conservées dans toutes les séquences 2019-nCoV disponibles analysées [Fig.S2, Fig.S3].
Les quatre inserts sont présents dans les génomes du virus aligné 28 Wuhan 2019-nCoV obtenus auprès de GISAID. L'écart dans le Bat-SARS Like CoV dans la dernière ligne montre que les insertions 1 et 4 sont très uniques à Wuhan 2019-nCoV.

Arbre phylogénétique de 28 isolats cliniques du génome de 2019-nCoV dont un provenant d'une chauve-souris en tant qu'hôte.
Nous avons ensuite traduit le génome aligné et constaté que ces inserts sont présents dans tous les virus Wuhan 2019-nCoV à l'exception du virus 2019-nCoV de chauve-souris en tant qu'hôte [Fig.S4]. Intrigués par les 4 inserts hautement conservés propres au 2019-nCoV, nous voulions comprendre leur origine. À cette fin, nous avons utilisé l'alignement local 2019-nCoV avec chaque insert comme requête contre tous les génomes viraux et pris en compte les hits avec une couverture de séquence de 100%. Étonnamment, chacun des quatre inserts est aligné avec de courts segments des protéines du virus de l'immunodéficience humaine-1 (VIH-1). Les positions des acides aminés des inserts dans 2019-nCoV et les résidus correspondants dans HIV-1 gp120 et HIV-1 Gag sont présentés dans le tableau 1. Les 3 premiers inserts (insert 1,2 et 3) alignés sur de courts segments d'acides aminés résidus dans le VIH-1 gp120. L'insert 4 aligné sur le VIH-1 Gag. L'insert 1 (6 résidus d'acides aminés) et l'insert 2 (6 résidus d'acides aminés) dans la glycoprotéine de pointe de 2019-nCoV sont 100% identiques aux résidus mappés à la gp120 du VIH-1. L'insert 3 (12 résidus d'acides aminés) dans 2019-nCoV correspond à la gp120 du VIH-1 avec des lacunes [voir le tableau 1]. L'insert 4 (8 résidus d'acides aminés) correspond au Gag du VIH-1 avec des lacunes.
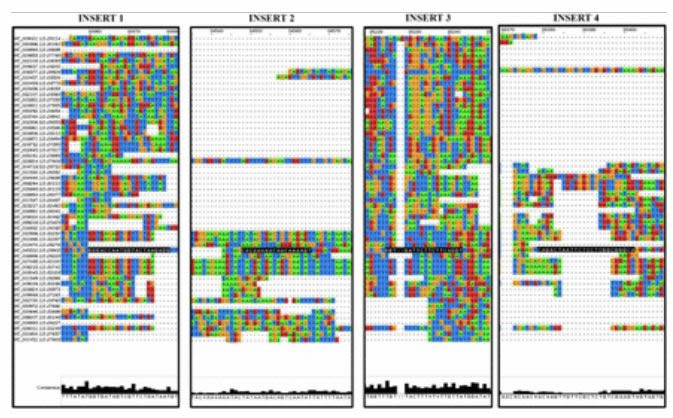
Alignement du génome de la famille des Coronaviridae. Les séquences noires surlignées sont les insertions représentées ici.
Tableau 1 :
Séquences alignées de 2019-nCoV et de la protéine gp120 du VIH-1 avec leurs positions dans la séquence primaire de la protéine. Tous les inserts ont une densité élevée de résidus chargés positivement. Les fragments supprimés dans les insertions 3 et 4 augmentent le rapport charge positive / surface. * veuillez consulter Supp. Tableau 1 pour les numéros d'accession
Bien que les 4 insertions représentent de courtes séquences d'acides aminés non contigus dans la glycoprotéine de pointe de 2019-nCoV, le fait que les trois partagent l'identité ou la similitude des acides aminés avec le VIH-1 gp120 et le VIH-1 Gag (parmi toutes les protéines virales annotées) suggère que ce n'est pas une conclusion fortuite aléatoire. En d'autres termes, on peut espérer sporadiquement une correspondance fortuite pour un tronçon de 6 à 12 résidus d'acides aminés contigus dans une protéine non apparentée. Cependant, il est peu probable que les 4 inserts de la glycoprotéine de pointe du nCoV 2019 correspondent fortuitement à 2 protéines structurelles clés d'un virus non apparenté (VIH-1).
Les résidus d'acides aminés des inserts 1, 2 et 3 de la glycoprotéine de pointe de nCoV 2019 qui ont été mappés sur le VIH-1 faisaient partie des domaines V4, V5 et V1 respectivement dans gp120 [Tableau 1]. Étant donné que les inserts 2019-nCoV mappés sur des régions variables du VIH-1, ils n'étaient pas omniprésents dans la gp120 du VIH-1, mais étaient limités à des séquences sélectionnées de VIH-1 [voir S.File1] provenant principalement d'Asie et d'Afrique.
La protéine VIH-1 Gag permet l'interaction du virus avec la surface hôte chargée négativement (Murakami, 2008) et une charge positive élevée sur la protéine Gag est une caractéristique clé de l'interaction hôte-virus. En analysant les valeurs de pI pour chacun des 4 inserts en 2019-nCoV et les séquences correspondantes de résidus d'acides aminés des protéines du VIH-1, nous avons constaté que :
a) les valeurs de pI étaient très similaires pour chaque paire analysée
b) la plupart de ces valeurs de pI étaient de 10 ± 2 [voir le tableau 1].
Il convient de noter que, malgré les lacunes dans les inserts 3 et 4, les valeurs pI étaient comparables. Cette uniformité dans les valeurs pI pour les 4 inserts mérite un examen plus approfondi.
Comme aucun de ces 4 inserts n'est présent dans aucun autre coronavirus, la région génomique codant pour ces inserts représente des candidats idéaux pour concevoir des amorces qui peuvent distinguer le 2019-nCoV des autres coronavirus.
Les nouveaux inserts font partie du site de liaison aux récepteurs de 2019-nCoV.
Pour obtenir des informations structurelles et comprendre le rôle de ces insertions dans la glycoprotéine 2019-nCoV, nous avons modélisé sa structure en fonction de la structure disponible de la glycoprotéine de pointe du SRAS (PDB: 6ACD.1.A). La comparaison de la structure modélisée révèle que bien que les inserts 1, 2 et 3 se trouvent à des emplacements non contigus dans la séquence primaire de la protéine, ils se replient pour constituer la partie du site de liaison de la glycoprotéine qui reconnaît le récepteur de l'hôte (Kirchdoerfer et al., 2016) (Figure 4). L'insert 1 correspond à la NTD (domaine N-terminal) et les inserts 2 et 3 correspondent au CTD (domaine C-terminal) de la sous-unité S1 dans la glycoprotéine de pointe 2019-nCoV. L'insert 4 est à la jonction des SD1 (sous-domaine 1) et SD2 (sous-domaine 2) de la sous-unité S1 (Ou et al., 2017). Nous supposons que ces insertions offrent une flexibilité supplémentaire au site de liaison de la glycoprotéine en formant une boucle hydrophile dans la structure protéique qui peut faciliter ou améliorer les interactions virus-hôte.
Analyse évolutive de 2019-nCoV
Il a été émis l'hypothèse que le 2019-nCoV est une variante du coronavirus dérivée d'une source animale qui a été transmise à l'homme. Compte tenu du changement de spécificité pour l'hôte, nous avons décidé d'étudier les séquences de glycoprotéines de pointe (protéine S) du virus. Les protéines S sont des protéines de surface qui aident le virus dans la reconnaissance et l'attachement de l'hôte. Ainsi, un changement dans ces protéines peut être reflété comme un changement de spécificité de l'hôte du virus. Pour connaître les altérations du gène de la protéine S de 2019-nCoV et ses conséquences dans les réarrangements structurels, nous avons effectué une analyse in-sillico de 2019-nCoV par rapport à tous les autres virus. Un alignement de séquences multiples entre les séquences d'acides aminés de la protéine S de 2019-nCoV, Bat-SARS-Like, SARS-GZ02 et MERS a révélé que la protéine S a évolué avec la diversité significative la plus proche du SARS-GZ02 (figure 1).Insertions dans la région de la protéine Spike de 2019-nCoV
Étant donné que la protéine S de 2019-nCoV partage l'ascendance la plus proche avec le SRAS GZ02, la séquence codant pour les protéines de pointe de ces deux virus a été comparée à l'aide du logiciel MultiAlin. Nous avons trouvé quatre nouvelles insertions dans la protéine de 2019-nCoV- «GTNGTKR» (IS1), «HKNNKS» (IS2), «GDSSSG» (IS3) et «QTNSPRRA» (IS4) (figure 2). À notre grande surprise, ces insertions de séquences étaient non seulement absentes dans la protéine S du SRAS mais n'ont également été observées chez aucun autre membre de la famille des Coronaviridae (figure supplémentaire). Cela est surprenant car il est peu probable qu'un virus ait acquis naturellement de telles insertions uniques en peu de temps.Les insertions partagent une similitude avec le VIH
Les insertions ont été observées dans toutes les séquences génomiques du virus 2019-nCoV disponibles dans les isolats cliniques récents (figure supplémentaire 1). Pour connaître la source de ces insertions dans 2019-nCoV, un alignement local a été effectué avec BLASTp en utilisant ces insertions comme requête avec tout le génome du virus. De façon inattendue, toutes les insertions se sont alignées avec le virus de l'immunodéficience humaine-1 (VIH-1). Une analyse plus approfondie a révélé que les séquences alignées de VIH-1 avec 2019-nCoV étaient dérivées de la glycoprotéine de surface gp120 (positions de séquence d'acides aminés: 404-409, 462-467, 136-150) et de la protéine Gag (366-384 acides aminés) ( Tableau 1). La protéine Gag du VIH est impliquée dans la liaison de la membrane de l'hôte, le conditionnement du virus et la formation de particules de type viral. Gp120 joue un rôle crucial dans la reconnaissance de la cellule hôte en se liant au récepteur primaire CD4. Cette liaison induit des réarrangements structurels dans GP120, créant un site de liaison de haute affinité pour un co-récepteur de chimiokine comme CXCR4 et / ou CCR5.Discussion
L'épidémie actuelle de 2019-nCoV justifie une enquête approfondie et une compréhension de sa capacité à infecter les êtres humains. En gardant à l'esprit qu'il y a eu un changement clair dans la préférence de l'hôte des coronavirus précédents à ce virus, nous avons étudié le changement de la protéine de pointe entre 2019-nCoV et d'autres virus. Nous avons trouvé quatre nouvelles insertions dans la protéine S de 2019-nCoV par rapport à son parent le plus proche, SARS CoV. La séquence du génome des 28 isolats cliniques récents a montré que la séquence codant pour ces insertions est conservée parmi tous ces isolats. Cela indique que ces insertions ont de préférence été acquises par le 2019-nCoV, ce qui lui confère un avantage supplémentaire de survie et d'infectivité.
En approfondissant, nous avons constaté que ces insertions étaient similaires au VIH-1. Nos résultats mettent en évidence une relation étonnante entre la gp120 et la protéine Gag du VIH, avec la glycoprotéine de pointe 2019-nCoV. Ces protéines sont essentielles pour que les virus puissent identifier leurs cellules hôtes et s'y accrocher et pour l'assemblage viral (Beniac et al., 2006). Puisque les protéines de surface sont responsables du tropisme de l'hôte, les changements dans ces protéines impliquent un changement dans la spécificité de l'hôte du virus. Selon des rapports de la Chine, il y a eu un gain de spécificité d'hôte dans le cas du 2019-nCoV, car le virus était à l'origine connu pour infecter les animaux et non les humains, mais après les mutations, il a également gagné le tropisme pour les humains.
À l'avenir, la modélisation 3D de la structure des protéines a montré que ces insertions sont présentes au site de liaison de 2019-nCoV. En raison de la présence de motifs gp120 dans la glycoprotéine de pointe du nCoV 2019 à son domaine de liaison, nous proposons que ces insertions de motifs auraient pu fournir une affinité accrue envers les récepteurs des cellules hôtes. De plus, ce changement structurel pourrait également avoir augmenté la gamme de cellules hôtes que le 2019-nCoV peut infecter. Au meilleur de nos connaissances, la fonction de ces motifs n'est pas encore claire dans le VIH et doit être explorée. L'échange de matériel génétique entre les virus est bien connu et cet échange critique met en évidence le risque et la nécessité d'étudier les relations entre des familles de virus apparemment sans rapport.
En approfondissant, nous avons constaté que ces insertions étaient similaires au VIH-1. Nos résultats mettent en évidence une relation étonnante entre la gp120 et la protéine Gag du VIH, avec la glycoprotéine de pointe 2019-nCoV. Ces protéines sont essentielles pour que les virus puissent identifier leurs cellules hôtes et s'y accrocher et pour l'assemblage viral (Beniac et al., 2006). Puisque les protéines de surface sont responsables du tropisme de l'hôte, les changements dans ces protéines impliquent un changement dans la spécificité de l'hôte du virus. Selon des rapports de la Chine, il y a eu un gain de spécificité d'hôte dans le cas du 2019-nCoV, car le virus était à l'origine connu pour infecter les animaux et non les humains, mais après les mutations, il a également gagné le tropisme pour les humains.
À l'avenir, la modélisation 3D de la structure des protéines a montré que ces insertions sont présentes au site de liaison de 2019-nCoV. En raison de la présence de motifs gp120 dans la glycoprotéine de pointe du nCoV 2019 à son domaine de liaison, nous proposons que ces insertions de motifs auraient pu fournir une affinité accrue envers les récepteurs des cellules hôtes. De plus, ce changement structurel pourrait également avoir augmenté la gamme de cellules hôtes que le 2019-nCoV peut infecter. Au meilleur de nos connaissances, la fonction de ces motifs n'est pas encore claire dans le VIH et doit être explorée. L'échange de matériel génétique entre les virus est bien connu et cet échange critique met en évidence le risque et la nécessité d'étudier les relations entre des familles de virus apparemment sans rapport.
Conclusions
Notre analyse de la glycoprotéine de pointe de 2019-nCoV a révélé plusieurs résultats intéressants :
Premièrement, nous avons identifié 4 inserts uniques dans la glycoprotéine de pointe de 2019-nCoV qui ne sont présents dans aucun autre coronavirus signalé jusqu'à ce jour. À notre grande surprise, les 4 insertions du nCoV 2019 ont été mappées à de courts segments d'acides aminés dans les gp120 et Gag du VIH-1 parmi toutes les protéines virales annotées de la base de données NCBI. Il est peu probable que cette similitude étrange de nouveaux inserts dans la protéine de pointe 2019-nCoV avec le VIH-1 gp120 et Gag soit fortuite.
De plus, la modélisation 3D suggère qu'au moins 3 des inserts uniques qui ne sont pas contigus dans la séquence protéique primaire de la glycoprotéine de pointe 2019-nCoV convergent pour constituer les composants clés du site de liaison du récepteur.
Il convient de noter que les 4 insertions ont toutes des valeurs de pI d'environ 10 qui peuvent faciliter les interactions virus-hôte. Pris ensemble, nos résultats suggèrent une évolution non conventionnelle de 2019-nCoV qui mérite une enquête plus approfondie. Nos travaux mettent en évidence de nouveaux aspects évolutifs du 2019-nCoV et ont des implications sur la pathogenèse et le diagnostic de ce virus.
Premièrement, nous avons identifié 4 inserts uniques dans la glycoprotéine de pointe de 2019-nCoV qui ne sont présents dans aucun autre coronavirus signalé jusqu'à ce jour. À notre grande surprise, les 4 insertions du nCoV 2019 ont été mappées à de courts segments d'acides aminés dans les gp120 et Gag du VIH-1 parmi toutes les protéines virales annotées de la base de données NCBI. Il est peu probable que cette similitude étrange de nouveaux inserts dans la protéine de pointe 2019-nCoV avec le VIH-1 gp120 et Gag soit fortuite.
De plus, la modélisation 3D suggère qu'au moins 3 des inserts uniques qui ne sont pas contigus dans la séquence protéique primaire de la glycoprotéine de pointe 2019-nCoV convergent pour constituer les composants clés du site de liaison du récepteur.
Il convient de noter que les 4 insertions ont toutes des valeurs de pI d'environ 10 qui peuvent faciliter les interactions virus-hôte. Pris ensemble, nos résultats suggèrent une évolution non conventionnelle de 2019-nCoV qui mérite une enquête plus approfondie. Nos travaux mettent en évidence de nouveaux aspects évolutifs du 2019-nCoV et ont des implications sur la pathogenèse et le diagnostic de ce virus.





{kind=link}